새로운 Senataxin 유전변이로 인한 눈돌림실행증동반실조 2형에서 나타난 체위반쪽시소안진
Positional Hemiseesaw Nystagmus in Ataxia with Oculomotor Apraxia Type 2 due to a Novel Senataxin Gene Mutation: A New Phenotype
Article information

Trans Abstract
Ataxia with ocular motor apraxia type 2 (AOA2) is an autosomal recessive disorder that is characterized by adolescent-onset gait ataxia, peripheral neuropathy, ocular apraxia, and cerebellar atrophy. A 19-year-old male with AOA2 from a novel SETX mutation showed distinct oculomotor abnormalities that included spontaneous and gaze-induced downbeat nystagmus, impaired smooth pursuit, and reversed catch-up saccades during horizontal head impulse tests, as well as peripheral neuropathy involving the lower extremities and mild slowing of frontal processing. He also showed positional hemiseesaw nystagmus in the supine and straight head-hanging positions. Positional hemiseesaw nystagmus is a new manifestation of hereditary cerebellar ataxia and may be explained by a gravity-dependent position-induced error in estimating the tilt in the roll plane due to dysfunction of the tilt-estimator circuit.
INTRODUCTION
Ataxia with ocular motor apraxia type 2 (AOA2) is an autosomal recessive disorder that is characterized by adolescentonset gait ataxia, peripheral neuropathy, ocular apraxia, and cerebellar atrophy [1]. Laboratory findings of AOA2 include elevated serum alpha-fetoprotein (AFP) and, less frequently, creatine kinase [1]. AOA2 results from recessive mutations of the senataxin gene (SETX) on chromosome 9q34 [2]. Herein we report positional hemiseesaw nystagmus (h-SSN), a new phenotype, in a patient with AOA2 due to a novel SETX mutation.
CASE REPORT
A 19-year-old Korean man presented with progressive ataxia for 4 years. He also reported dysarthria, tremor of the hands, painless numbness of both legs, and intermittent diplopia. He had no family history of neurologic disorders. Neurologic examination found esophoria on alternating cover tests, normal range of ocular motion, and downbeat nystagmus (DBN) during lateral gazes and after head-shaking. In darkness, the patient showed subtle DBN just after blinking. Horizontal saccades were normal. Smooth pursuit was impaired bilaterally. Immediately after lying down or straight head hanging, DBN increased for about 3 seconds, and then changed into h-SSN (Supplementary video clip). The fast components of h-SSN were upward in the right eye and downward in the left eye, and counterclockwise (from the patient’s perspective) in both eyes. The h-SSN was maintained throughout the lying down position lasting 2 minutes. Rolling his head to either side while supine increased DBN and induced apogeotropic nystagmus, stronger during leftward head turning. Reversed corrective saccades were observed during head impulse tests (HITs) for both horizontal canals. He also showed impaired tandem gait, falling to either side while attempted tandem Romberg testing, decreased sensation in both legs, absent knee tendon reflexes, and high-arched feet. There was no ocular motor apraxia.
Neuropsychological evaluations revealed slow responses in the Stroop test and impaired time scores in the Trail Making Test-B, but with normal attention, memory, visuospatial construction, and intelligence. Nerve conduction studies found axonal sensorimotor neuropathy of both legs. Brain magnetic resonance imagings (MRIs) showed cerebellar atrophy mainly involving the rostral vermis (Fig. 1A, B). Serum AFP was increased at 14.5 ng/mL (normal <8.1 ng/mL), but cobalamin, vitamin E, lactate, creatine kinase, and lipid profiles were normal. Genetic tests for Friedreich’s ataxia and spinocerebellar ataxia were negative. Further molecular genetic analyses were performed using the next-generation sequencing technology after informed consent. After filtering out the variants which were detected in more than 1% among the control population using the public (1000 Genomes Project, Exome Variant Server, and Exome Aggregation Consortium) and in-house database, 2 new variants were identified in SETX; NM_015046.5:c.1738G>A (p.Glu580Lys) and NM_015046.5:c.5088dupT (p.Val1697Cysfs*3) (Fig. 1C, D). The c.5088dupT mutation was considered pathogenic since it is a frameshift variant leading to a premature protein truncation.
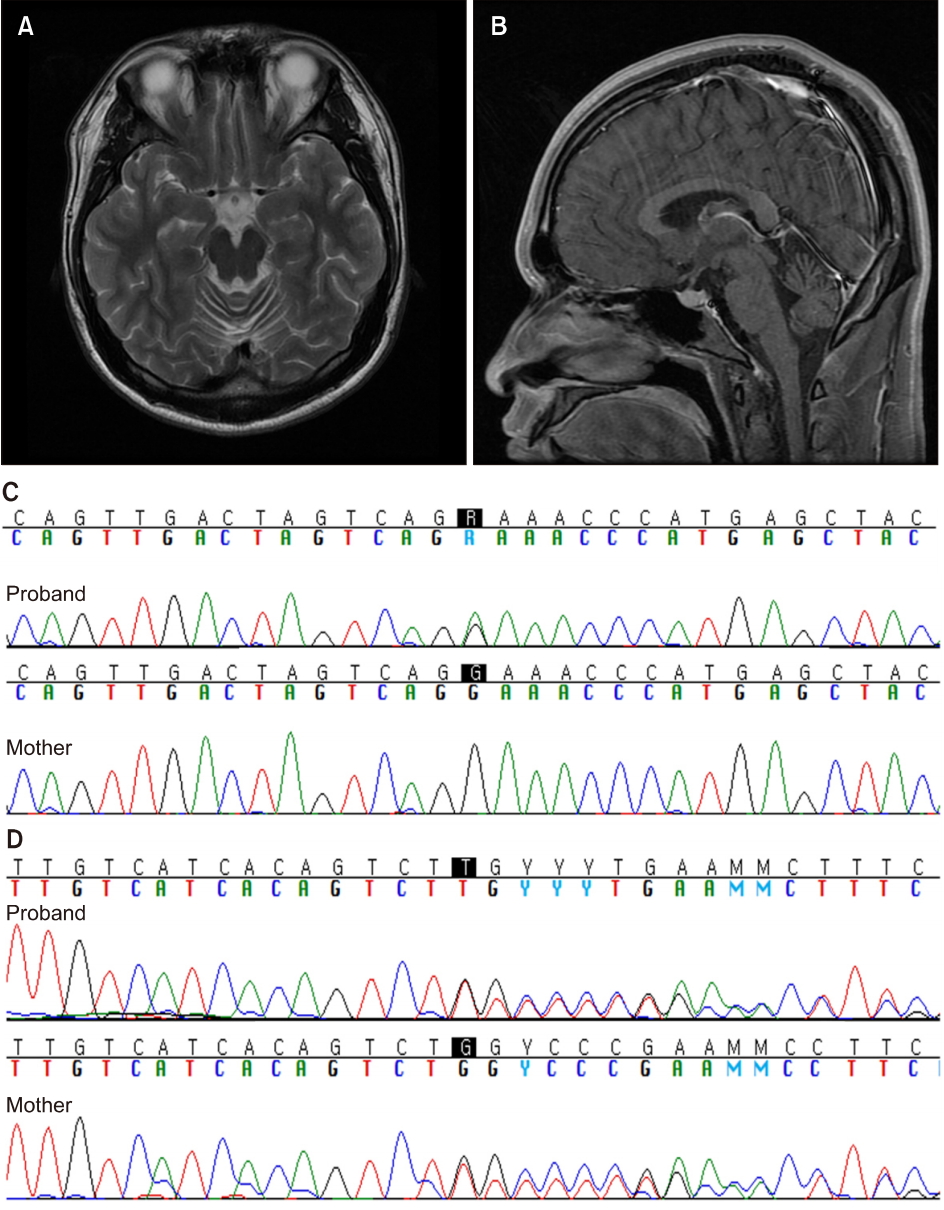
(A, B) Brain magnetic resonance imaging shows cerebellar atrophy, more marked in the rostral vermis. (C) Sequencing chromatogram of the SETX variants detects a c.1738G>A variant in the proband, but not in his mother. (D) A c.5088dupT variant inherited from his mother was also detected.
DISCUSSION
In addition to peripheral neuropathy involving the lower extremities and mild slowing of frontal processing, our patient with AOA2 from novel genetic defects in SETX showed ocular motor abnormalities such as spontaneous and gaze-induced DBN, impaired smooth pursuit, and reversed catch-up saccades during horizontal HITs. He also showed positional h-SSN in the supine and straight head-hanging positions that have not been described in the literature. AOA2 belongs to the autosomal recessive cerebellar ataxia that is characterized by degeneration of the cerebellum, spinal cord, and often peripheral nerves. Although it is included in the acronym, ocular motor apraxia is not a mandatory finding in AOA2, likewise in our patient. Ocular motor apraxia was found in 20% of Italian patients [1], in 32% of Algerian patients [3], and in none of the Quebec cluster [4].
AOA2 arises from mutations in the SETX encoding senataxin, a DNA/RNA helicase. Senataxin seems to be involved in the defense against DNA damage, and to play an important role in survival of the cerebellar, anterior horn, and dorsal root neurons [1]. Cerebellar atrophy is an early finding on MRIs, observed in more than 90% of AOA2 patients [1-3]. A postmortem study of AOA2 revealed a loss of Purkinje cells and fibrous gliosis, more severe in the vermis than in the hemispheres [3]. Dysfunction of the vestibulocerebellum involving the flocculus may explain majority of the ocular motor abnormalities observed in our patient, including spontaneous and head-shaking DBN, and gaze-holding deficits [5]. Since the vestibulocerebellum concerns the vestibulo-ocular reflex gain through inhibitory projections to the vestibular nuclei, disinhibited vestibular nuclei may lead to increased vestibulo-ocular reflex gain and reversed corrective saccades during HITs [6].
The h-SSN refers to a jerk-waveform nystagmus that has conjugate torsional but disjunctive vertical components. Since h-SSN in the head erect usually occurs in unilateral brainstem lesions, imbalance of the vestibular pathways from the vertical canals or the otolithic organs has been implicated in generation of h-SSN [7]. Whereas DBN increased immediately after straight lying down and head hanging, maintaining those positions resulted in evolution into h-SSN in our patient. The paroxysmal DBN upon straight lying down and head hanging may be explained by excessive postacceleratory inhibition of the posterior canals during the positioning due to vestibulocerebellar dysfunction [8]. In contrast, the following h-SSN maintained throughout the supine position may be explained by a gravity-dependent position-induced error in estimating the tilt in the roll plane due to dysfunction of the tilt-estimator circuit in which information from the semicircular canals about head rotation is combined with otolith information about linear acceleration through the velocity-storage mechanism [9].
Notes
No potential conflict of interest relevant to this article was reported.
SUPPLEMENTARY MATERIAL
Immediately after lying down or straight head hanging, downbeat nystagmus in darkness increased for about 3 seconds, and then changed into hemiseesaw nystagmus.
Supplementary video clip can be found via https://doi.org/10.21790/rvs.2020.19.1.12
rvs-19-1-12-suppl.mp4